
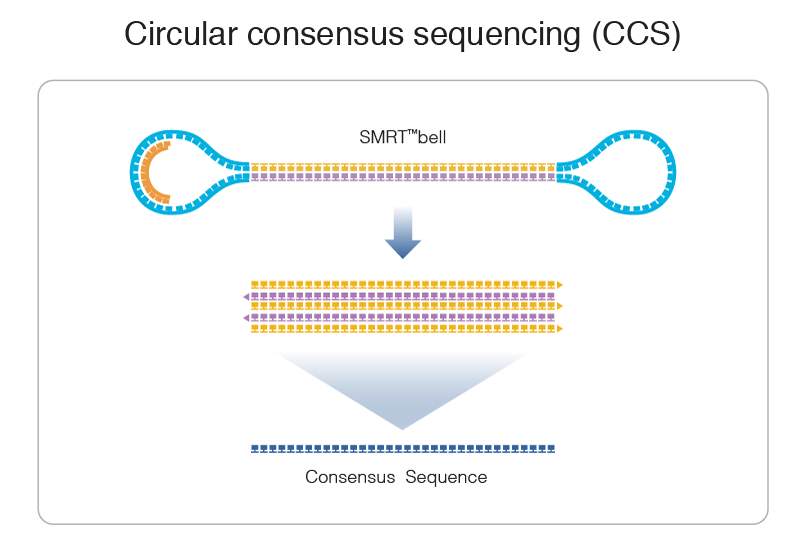
The ccs program takes multiple reads of the same SMRTbell sequence and combines them to produce one high quality consensus sequence.
bax2bam -o mynewbam mydata.1.bax.h5 mydata.2.bax.h5 mydata.3.bax.h5
ccs myresults.bam mynewbam.subreads.bam
The ccs program needs a .subreads.bam file containing the subreads for each SMRTbell sequenced. Older versions of the PacBio RS software outputted data in bas.h5 files, while the new software outputs BAM files. If you have a bas.h5 file from the older software, you will need to convert it into a BAM. This can be done with the tool bax2bam which simply needs the name of any bas.h5 files to convert and the prefix of the output file. Assuming your original file is named mydata.bas.h5, you can produce a file mynewbam.subreads.bam with the following command.
bax2bam -o mynewbam mydata.1.bax.h5 mydata.2.bax.h5 mydata.3.bax.h5
CCS can be run at the command line as follows:
ccs [OPTIONS] OUTPUT FILES...
An example command would be:
ccs --minLength=100 myCCS.bam myData.subreads.bam
The CCS program can be run with a variety of options described below.
| Option | Example (Defaults) | Explanation |
|---|---|---|
| Output File Name | myResult.bam | This argument is the first argument that comes after the named arguments shown below. |
| Input Files | myData.subreads.bam | The end of the CCS command following the named arguments and the output file name is the name of one, or more, subread.bam files to be processed. |
| Version Number | --version | Prints the version number |
| Report File Name | --reportFile=ccs_report.txt | The report file contains a result tally of the outcomes for all ZMWs that were processed. If the filename is not given the report is outputted to the text file ccs_report.txt In addition to the count of successfully produced consensus sequences, this file lists how many ZMWs failed various data quality filters (SNR too low, not enough full passes, etc.) and is useful for diagnosing unexpected drops in yield. |
| Minimum SNR | --minSnr=3.75 | This filter removes data that is likely to contain deletions. SNR is a measure of the strength of signal for all 4 channels (A, C, G, T) used to detect basepair incorporation. The SNR can vary depending on where in the ZMW a SMRTbell stochastochastically lands when loading occurs. SMRTbells that land near the edge and away from the center of the ZMW have a less intense signal, and as a result can contain sequences with more "missed" basepairs. This value sets the threshold for minimum required SNR for any of the four channels. Data with SNR < 3.75 is typically considered lower quality. |
| Minimum Read Score | ----minReadScore=0.75 | Sets a minimum value for the predicted quality of any subread used for CCS. Note that this filters the input to CCS (the subread quality must be above this value), whereas the --minPredictedAccuracy option filters the output (the predicted consensus sequence must be above a certain predicted accuracy). |
| Minimum Length | --minLength=10 | Sets a minimum length requirement for the median size of insert reads in order to generate a consensus sequence. If the targeted template is known to be a particular size range, this can filter out alternative DNA templates. |
| Maximum Length | --maxLength=7000 | Sets a maximum length for both subreads that will be processed as well as the consensus sequence that will be generated. For robust results while avoiding unnecessary computation on unusual data, this can be set to ~20% above the largest expected insert size. |
| Minimum Number of Passes | --minPasses=3 | Sets a minimum number of passes for a ZMW to be emitted. This is the number of full passes. Full passes must have an adapter hit before and after the insert sequence and so does not include any partial passes at the start and end of the sequencing reaction. Additionally, the full pass count does not include any reads that were dropped by the Z-Filter. |
| Minimum Predicted Accuracy | --minPredictedAccuracy=0.9 | The minimum predicted accuracy of a read. CCS generates an accuracy prediction for each read, defined as the expected percentage of matches in an alignment of the consensus sequence to the true read. A value of 0.99 indicates that only reads expected to be 99% accurate are emitted. |
| Minimum Z Score | --minZScore=-3.5 | The minimum Z-Score for a subread to be included in the consensus generating process. For more information, see the "What are Z-Scores" section below. |
| Maximum Dropped Fraction | --maxDropFraction=0.34 | The maximum number of subreads that can be dropped before the entire ZMW is discarded. Subreads that appear very unlikely given the initial template (low Z-score), are discarded before generating the consensus sequence as part of an initial quality filter. Typically, very few reads should be discarded but if a high proportion are, then the entire ZMW is dropped. |
| ZMWs to Process | --zmws=0-100000000000 | If the consensus sequence for only a subset of ZMWs is required, they can be specified here. ZMWs an be specified either by range (--zmws=1-2000) by values (--zmws=5,10,20), or by both (--zmws=5-10,35,1000-2000). Simply use a comma separated list with no spaces. |
| Number of Threads to Use | --numThreads=0 | How many threads to use while processing. By default, ccs will use as many threads as there are available cores to minimize processing time, but fewer threads can be specified here. |
| Log File | --logFile=mylog.txt | The name of a log file to use, if none is given the logging information is printed to STDERR. |
| Log level verbosity | --logLevel=INFO | How much log data to produce? By setting --logLevel=DEBUG, you can obtain detailed information on what ZMWs were dropped during processing, as well as any errors which may have appeared. |
| Disable Polishing | --noPolish | After constructing the initial template, do not proceed with the polishing steps. This is significantly faster, but generates less accurate data with no RQ or QUAL values associated with each base. |
| Analyze strands separately | --byStrand | Separately generate a consensus sequence from the forward and reverse strands. Useful for identifying heteroduplexes formed during sample preparation. |
| Overwrite output file | --force | When you don't care it already exists. |
When the ccs program finishes, it outputs a BAM file with one entry for each consensus sequence derived from a ZMW. BAM is a general file format for storing sequence data, which is described fully by the SAM/BAM working group here. The CCS output format is a version of this general format, where the consensus sequence is represented by the "Query Sequence" and several tags have been added to provide additional meta information.
An example BAM entry for a consensus as seen by samtools is shown below.
m141008_060349_42194_c100704972550000001823137703241586_s1_p0/63/ccs 4 * 0 255 * * 0 0 CCCGGGGATCCTCTAGAATGC ~~~~~~~~~~~~~~~~~~~~~ RG:Z:83ba013f np:i:35 rq:i:999 rs:B:i,37,0,0,1,0 sn:B:f,11.3175,6.64119,11.6261,14.5199 za:f:2.25461 zm:i:63 zs:B:f,-1.57799,3.41424,2.96088,2.76274,3.65339,2.89414,2.446,3.04751,2.35529,3.65944,2.76774,4.119,1.67981,1.66385,3.59421,2.32752,4.17803,-0.00353378,nan,0.531571,2.21918,3.88627,-0.382997,0.650671,3.28113,0.798569,4.052,0.933297,3.00698,2.87132,2.66324,0.160431,1.99552,1.69354,1.90644,1.64448,3.13003,1.19977
A description of each of the common separated fields is given below.
| Name | Definition |
|---|---|
| Query Name | Movie Name / ZMW # /ccs |
| FLAG | Required by format but meaningless in this context. Always set to indicate the read is unmapped (4) |
| Reference Name | Required by format but meaningless in this context. Always set to '*' |
| Mapping Start | Required by format but meaningless in this context. Always set to '0' |
| Mapping Quality | Required by format but meaningless in this context. Always set to '255' |
| CIGAR | Required by format but meaningless in this context. Always set to '*' |
| RNEXT | Required by format but meaningless in this context. Always set to '*' |
| PNEXT | Required by format but meaningless in this context. Always set to '0' |
| TLEN | Required by format but meaningless in this context. Always set to '0' |
| Consensus Sequence | This is the consensus sequence generated. |
| Quality Values | This is the per-base parameteric quality metric. For more information see the "interpretting QUALs" section. |
| RG Tag | This is the read group identifier. |
| bc Tag | This is a 2-entry array of upstream-provided barcode calls for this ZMW. |
| bq Tag | This is the quality of the barcode call. (optional, depends on barcoded inputs) |
| np Tag | The number of full passes that went into the subread. (optional, depends on barcoded inputs) |
| rq Tag | The predicted read quality. |
| rs Tag | An array of counts for the effect of adding each subread. The first element indicates the number of success and the remaining indicate the number of failures. This is a comma separated list of the number of reads Successfully Added, Failed to Converge in likelihood, Failed the Z Filtering, Failed to pass the pre-POA size filtering, or were excluded for another reason. |
| za Tag | The average Z-score for all reads successfully added. |
| zm Tag | The ZMW hole number. |
| zs Tag | This is a comma separated list of the Z-scores for each subread when compared to the initial candidate template. A "nan" value indicates that the subread was not added. |
The QUAL value of a read is a measure of the posterior likelihood of an error at a particular position. Increasing QUAL values are associated with a decreasing probability of error. For the case of Indels and homopolymers, it is there is ambiguity as to which QUAL value is associated with the error probability. Shown below are different types of alignment errors, with a '*' indicating which sequence BP should be associated with the alignment error.
*
ccs: ACGTATA
ref: ACATATA
*
ccs: AC-TATA
ref: ACATATA
*
ccs: ACGTATA
ref: AC-TATA
Indels should always be left-aligned and the error probability is only given for the first base in a homopolymer.
* *
ccs: ACGGGGTATA ccs: AC-GGGTATA
ref: AC-GGGTATA ref: ACGGGGTATA
Z-score filtering is a way to remove outliers and contaminating data from the CCS dataset prior to consensus generation, a crucial step for any analysis. For example, in the second world war the U.S. Navy tried to gauge the accuracy of a newly developed optical range finder by having a few hundred sailors practice on a known target. The mean accuracy was quite poor, but after realizing that 20% of people cannot view sterotypically, these individuals could be excluded, leading to much higher overall accuracy.
The Z-score for a subread is a metric which quantifies how well it doesn't fit the model or assumptions of CCS scoring. In CCS, an initial template sequence is proposed, and then further refined using data in the templates. The initial template is usually quite close to the final consensus sequence, and at this stage ccs will evaluate how likely each read is based on the candidate template. The likelihood of a read for a template is summarized by it's Z-score, which asymptotically is normally distributed with a mean near 0.
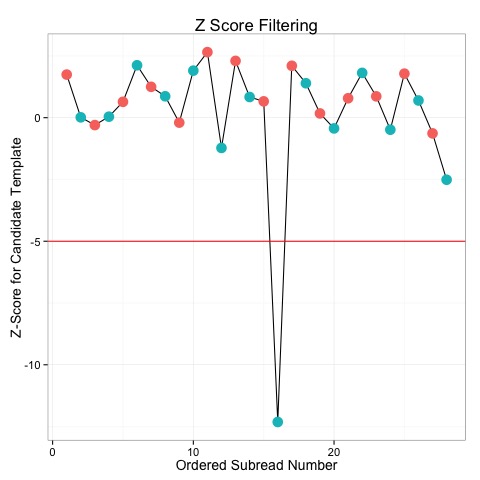
Subreads with very low Z-scores are very unlikely to have been produced according to the CCS model, and so represent outliers. For example, the plot below shows the Z-scores for several subreads. With a -5 cutoff, we can see that one subread is excluded from the data.
After ccs runs, a report is generated that specifies the number of ZMWs that successfully produced consensus sequences, as well as a count of how many ZMWs did not produce a consensus sequence for various reasons. The entries in this report, as well as parameters that can be set to change to increase of decrease the number of ZMWs that pass various filters, are explained in the table below.
| ZMW result | Parameter(s) affecting result | Description |
|---|---|---|
| Below SNR threshold | --minSnr | ZMW had at least one channel's SNR below the minimum threshold. |
| No usable subreads | --minReadScore, --minLength, --maxLength | The ZMW had no usable subreads, either there were no subreads, or all the subreads were below the minimum quality threshold or were above/below the specified length thresholds. |
| Insert size too long | --maxLength | The consensus sequence was above the maximum length threshold. Note that if all the input subreads were already above this threshold, they would all have been excluded leading to "No usable subreads." |
| Insert size too small | --minLength | The consensus sequence was below the miminum length threshold. Note that if all the input subreads were already below this threshold, they would all have been excluded leading to "No usable subreads." |
| Not enough full passes | --minPasses | There were not enough subreads that had an adapter at the start and end of the subread (a "full pass"). |
| Too many unusable subreads | --minZScore, --maxDropFraction | The ZMW had too many subreads that could not be used. A read can be unusable if it appears too unlikely given the initial template (low Z-score), or rarely, if a numerical rounding error occurs during processing. |
| CCS did not converge | None | The consensus sequence did not converge after the maximum number of allowed rounds of polishing. |
| CCS below minimum predicted accuracy | --minPredictedAccuracy | Each CCS read has a predicted level of accuracy associated with it, reads that are below the minimum specified threshold are removed. |
| Unknown error during processing | None | These should not occur. |