gmx2qmmm is a python interface for Quantum mechanics/Molecular mechanics (QM/MM) calculation.
- Overview
- System requirments
- gmx2qmmm job
- Input files
- Examples
- References
- Support and development
- Links
gmx2qmmm is a python package to bridge Gaussian and Gromacs. The test runs were performed using Gaussian16 and Gromacs 5.0.2, but the code should be able to read earlier Gaussian and other Gromacs versions. The only limits are the formats of the human-readable input and output files of each program, as such, conversion scripts can be written to make the interface work with any version, if the current code does not support it.
Conceptually, gmx2qmmm creates a QM/MM potential and performs either single point calculations (i.e., the current energy of your system), geometry optimizations, and linear relaxed scan. (Other ultilities are ongoing)
- python 3.6+
- Gaussian16 (and earlier version)
- Gromacs 5.0.2 (and earlier version)
| Job type | Calculation |
|---|---|
| Single point calcuation (SP) | Calculate single point energy and forces (.xyz) |
| Geometry optimizations (OPT) | Optimize the system energy via optimizer (Steepest descent, Conjugate gradient or BFGS) |
| Relaxed Scan (SCAN) | Relaxed linear scan (angle and dihedral angle are in development) |
The required input files in the package are
| Input files | Command | Default input name |
|---|---|---|
| Coordinate file (.g96 or .gro) | -c | conf.g96 |
| Topology (.top) | -p | topol.top |
| QM atoms file(.ndx) | -n | qmatoms.ndx |
| QM parameters (.dat) | -qm | qm.dat |
| MM parameters (.dat) | -mm | mm.dat |
| QM/MM parameters (.dat) | -qmmm | qmmm.dat |
| Active atoms (.ndx) | -act | act.ndx |
| Path file (.dat) | -path | path.dat |
| Logfile (.log) | -g | logfile |
If the parameters are not entered in the input files, the program will run the job with the default values.
Input files guide please execute python gmx2qmmm.py -h
The executed example with default parameters is python gmx2qmmm.py
For advance information please read the Documentation.
The directory example contains SP, OPT, linear SCAN calculation for glycine serine (GLYSER).
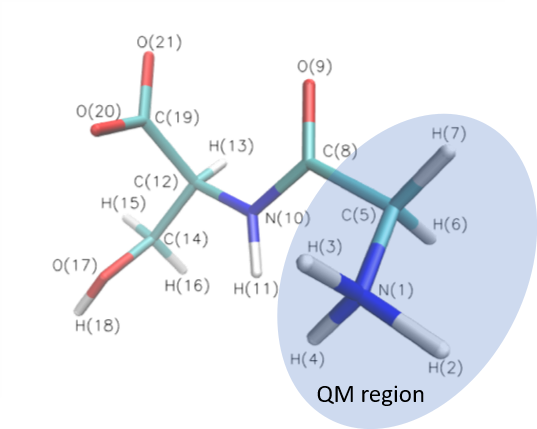
The names of the input files are default. Go to the sp/opt directory on the command line and run:
python gmx2qmmm.py
A user‐friendly, Python‐based quantum mechanics/Gromacs interface: gmx2qmmm Jan P. Götze, Yuan‐Wei Pi, Simon Petry, Fabian Langkabel, Jan Felix Witte, Oliver Lemke https://doi.org/10.1002/qua.26486
For bug reports/suggestions/complaints please raise an issue on GitHub.
Or contact us directly: gmx2qmmm@gmail.com