Fastq files demultiplexer, handling double indexing, molecular indexing and filtering based on index quality (Pure Python2.7)
Creation : 2015/01/07
Last update : 2016/11/21
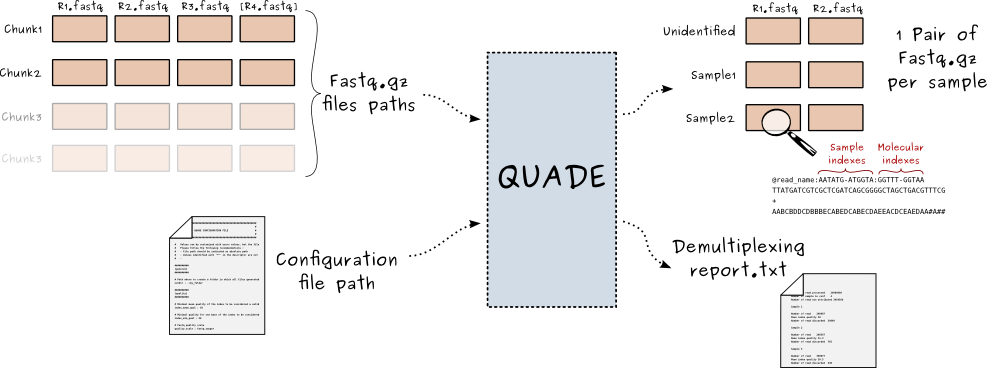
Quade is a python2.7 object oriented script developed to demultiplex sample from mixed fastq files.
Specific features:
- The program parse chunks of non demultiplexed raw paired end fastq files (converted in fastq by CASAVA but not demultiplexed, see convert-bcl-to-fastq)
- Reads are attributed to a specific sample defined by index sequences. Simple and double indexing are supported.
- Reads can be filtered to exclude index read with bad quality base (to avoid sample cross contamination due to errors in index read)
- The program handle stochastic molecular index. Sample index and molecular index are appended at the end of sequence name for further use.

- A configuration file containing all program parameters (including sample/index association) is parsed and thoroughly verified for validity
- Non demultiplexed fastq files are read with a custom fastq parser (pyFastq Submodule) chunks by chunks, with reads from sample read1, sample read2, index read1 [and index read2] in different files.
- sample barcode and molecular index sequence are extracted from index read. In the case of double indexing sample barcode is the result of the fusion between sample barcode from index1 and index2. The same applies for molecular index
- Depending of the index (or fused index) sequence, sample reads1 and read2 are attributed to a defined sample, or to the undetermined category if appropriate.
- The phred quality of the index read is checked so as every positions is above a defined minimal value. If so reads are written in a sample pass file, else reads are written in a sample fail file.
- reads names are written as follow : @Name_in_original_fastq:SAMPLE_BARCODE[:MOLECULAR_BARCODE]
- a distribution report is generated
The program was developed under Linux Mint 17 and was not tested with other OS. In addition to python2.7 the following dependencies are required for proper program execution:
- python package numpy 1.7.1+
If you have pip already installed, enter the following line to install packages: sudo pip install numpy HTSeq
-
Clone the repository in recursive mode to download the main repo and its submodules
git clone --recursive https://github.com/a-slide/Quade.git -
Enter the src folder of the program folder and make the main script executable
sudo chmod u+x Quade.py -
Finally, add Quade.py to your PATH
In the folder where fastq files will be created
Usage: Quade.py -c Conf.txt [-i -h]
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-c CONF_FILE Path to the configuration file [Mandatory]
-i Generate an example configuration file and exit [Facultative]
An example configuration file can be generated by running the program with the option -i The possible options are extensively described in the configuration file. The program can be tested from the test folder with the dataset provided and the default configuration file.
cd ./test/result
Quade.py -i
Quade.py -c Quade_conf_file.txt
See Blog post Starting a new NGS project with python : Example with a fastq demultiplexer
- Adrien Leger aleg@ebi.ac.uk @a-slide
- Emilie Lecomte emilie.lecomte@univ-nantes.fr @emlec