



orfipy is a tool written in python/cython to extract ORFs in extremely fast and flexible manner. Please read the preprint here
pip install orfipy
Or install via conda
conda install -c bioconda orfipy
git clone https://github.com/urmi-21/orfipy.git
cd orfipy
pip install .
or use pip
pip install git+git://github.com/urmi-21/orfipy.git
Details of orfipy algorithm are in the preprint and SI. Please go through the SI if you are interested to know differences between orfipy and other ORF finder tools and how to set orfipy parameters to match the output of other tools.
Below are some usage examples for orfipy
To see full list of options use the command:
orfipy -h
orfipy currently supports sequences in only Fasta format. If you want to directly work with compressed Fasta files, compression format must be bgzip. To use Fastq files, you will need to first convert them to Fasta.
Extract ORF sequences and write ORF sequences in orfs.fa file
orfipy input.fasta --dna orfs.fa --min 10 --max 10000 --procs 4 --table 1 --outdir orfs_out
Use standard codon table but use only ATG as start codon
orfipy input.fasta --dna orfs.fa --start ATG
Note: Users can also provide their own translation table, as a .json file, to orfipy using --table option. Example of json file containing a valid translation table is here
See available codon tables
orfipy --show-table
Extract ORFs BED file
orfipy input.fasta --bed orfs.bed --min 50 --procs 4
or
orfipy input.fasta --min 50 --procs 4 > orfs.bed
Extract ORFs BED12 file
Note: Add --include-stop for orfipy output to be consistent with Transdecoder.Predict output .bed file.
orfipy testseq.fa --min 100 --bed12 of.bed --partial-5 --partial-3 --include-stop
Extract ORFs peptide sequences using default translation table
orfipy input.fasta --pep orfs_peptides.fa --min 50 --procs 4
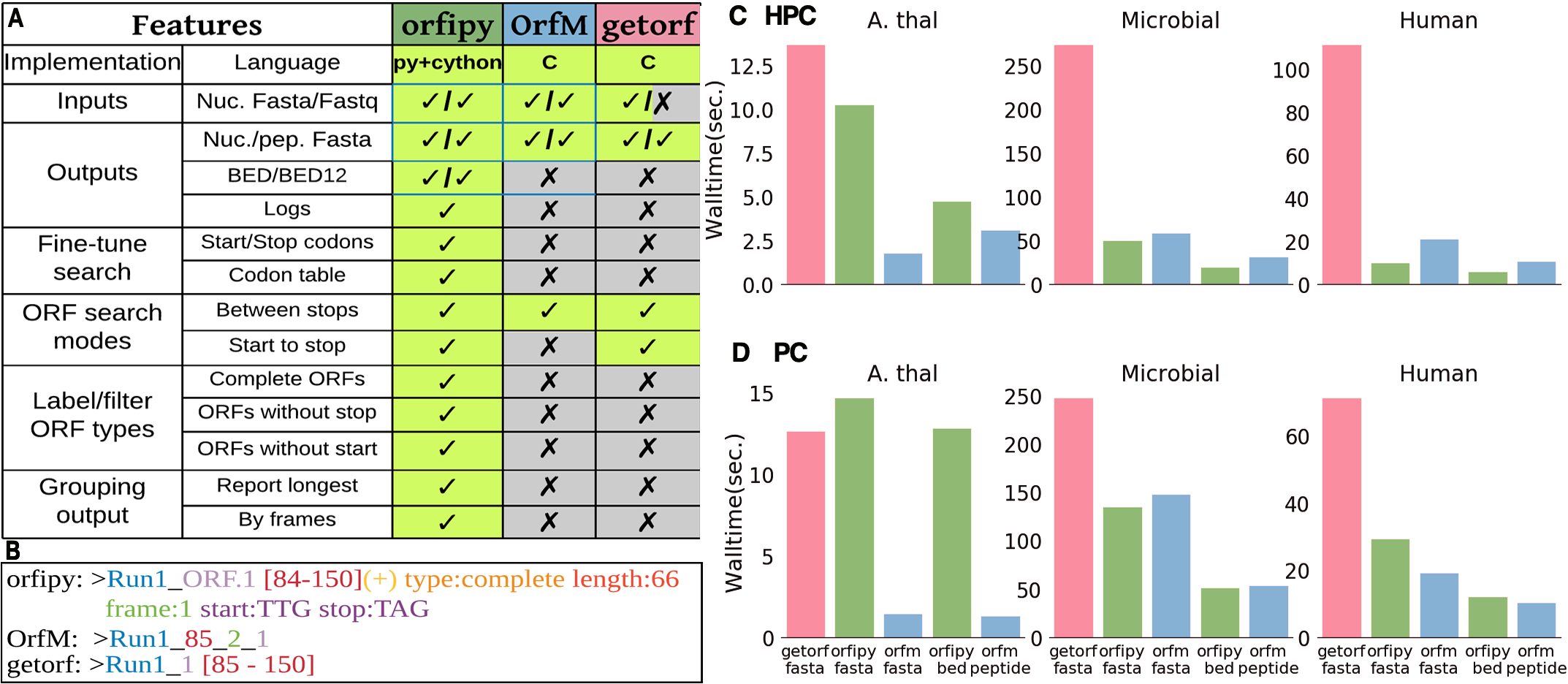 Comparison of orfipy features and performance with getorf and OrfM. For details see preprint and SI
Comparison of orfipy features and performance with getorf and OrfM. For details see preprint and SI