{kind=link}
Mpipe is a Bioinformatics tool for compare DNA motif occurrence at peak summits versus surrounding regions.
This program involves three major functions:
- Given regions in the genome, fetch DNA sequences.
- Use a motif PSSM to scan the 200bp DNA sequence and calculate a score for the motif occurrence.
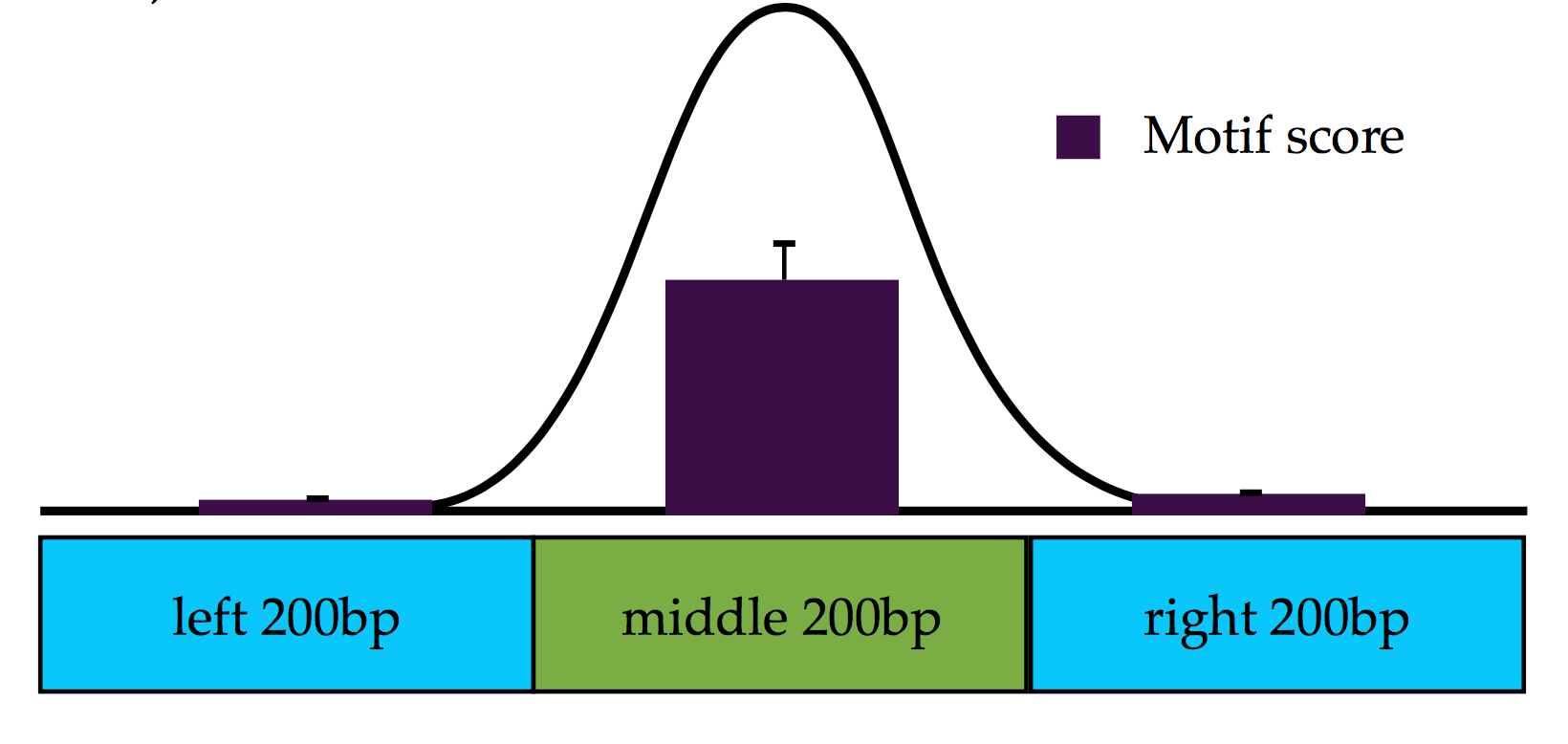
- Calculate a statistical measurement to see higher scores in middle 200bp windows than two surrounding windows. Measure the difference of summary scores distributions in middle v.s two surrounding windows.
BioPython is used to input fasta sequence file. (http://biopython.org/wiki/Download)
Bed tools is used to convert bed to fasta file (fastaFromBed)and sort bed by its position (bedSort). (http://code.google.com/p/bedtools/) (Download bedSort by: git clone http://genome-source.cse.ucsc.edu/kent.git)
RPy is used to do the Wicoxon test and FDR adjust between center region and side regions. (http://rpy.sourceforge.net/rpy.html)
awk is used to get three regions of bed from one summits bed and to check whether a region is in legal chromosome range (gawk in Linux)
Cython is only needed for developers. (http://cython.org/)
Python 2.6 or above is recommended.
You can download the newest non-installed package by typing in command:
$ git clone https://github.org/hanfeisun/Mpipe
$ python setup.py install
Run the whole pipeline for first time:
$ Mpipe.py -b summits.bed -m motif_database.xml -g
../assembly/human19/masked -o P300
After the first run, a fasta file named by 'hg19.fa' will be generated, which can be used directly in the later runs:
$ Mpipe.py -b summits.bed -m motif_database.xml -g
hg19.fa -o P300
There are alse two small tools in these package, to test the integrity of a xml file and view it:
$ motif_xml_view.py motif.xml
To test the integrity of a fasta file and view its GC content:
$ seq_GC_view.py hg19.fa
You can see other usage by:
$ Mpipe.py -h
17 files will be output into ONE directory
They are:
- fasta file and bed file for center region and side regions.
- pickle file and txt file with scores of every peak for center region and side regions.
- html file with scores of every peak in center region. (CAUTION: opening this html file is memory costing, need more than 1G memory for 3000 peaks and 700 motifs)
- html file and txt file with summary scores of every motif
- a pdf file show the distribution of p-value and difference of mean
- the original summits bed file
The most useful outputs are:
-
PvM (Peaks vs Motifs) table, which is named by prefix_middle.txt (Example: https://hanfeisun.github.io/Mpipe_output/P300/P300_middle.txt)
-
MSM (Motif Score Metric) table, which is named by prefix_metric.txt (Example: https://hanfeisun.github.io/Mpipe_output/P300/P300_metric.txt)
-
MSMC (Motif Score Metric Colored) table, which is named by prefix_metric.html, colored by its pvalue and the difference of mean between center and side regions. (Example: https://hanfeisun.github.io/Mpipe_output/P300/P300_metric.html)
-
The Histogram of difference of means between center and two sides (Example: https://hanfeisun.github.io/Mpipe_output/P300/P300_dist.pdf)