This is a project I worked on while in university. There are two main components, the first is code to read molecular geometry files as well as visualize these in 3d. The second part performs Hartree-Fock calculations. Hartree-Fock is a “method of approximation for the determination of the wave function and the energy of a quantum many-body system in a stationary state”. I became interested in running these calculations to learn more about what properties of a molecule could be computed , ie ground state energy or molecular structure.
The molecular geometry reads molecule files in both Z-matrix format and XYZ format. There is also support for two visualization modes: Vpython and vispy. The vpython implementation is faster for larger molecules, although a very good (and scalable) vispy molecular viewer can be found in the vispy examples directory.
Usage: python visualize_molecule.py [method] molecule_file
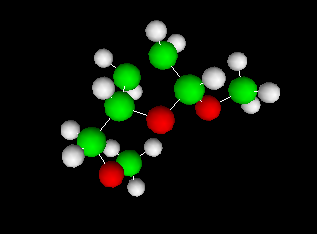
Here are some examples of the visualizer (all with vpython mode):

The Hartree-Fock code can perform restricted closed shell HF calculations, also known as the Self Consistent Field method (SCF). These calculations allow you to compute the energy of a molecule in different configurations. I based most of the work of how to implement this on the first few chapters of the excellent book Modern Quantum Chemistry.
There are a few components: The program can read molecular basis sets. These consist of coefficients and exponents of Gaussian type orbitals which approximate Slater orbitals and make the calculations easier.
After reading molecular geometry and the chosen basis set, a number of different matricies are calculated as well as the two electron repulsion integrals. This is by far the most expensive part of a SCF calculation, scaling as O(N^4) where N is the number of basis functions required to represent the atomic orbitals.
This implementation can perform a restricted set of calculations on S-type orbitals only. Furthermore, it is implemented in a fairly inefficient manner - all numerical code in python - as the goal of this project was to understand the mechanics of the SCF procedure. Once these calculations are complete, an iterative convergence is performed which computes the final coefficient matrix.
Usage: python compute_hartree_fock.py [molecule_file] [basis_set_file]
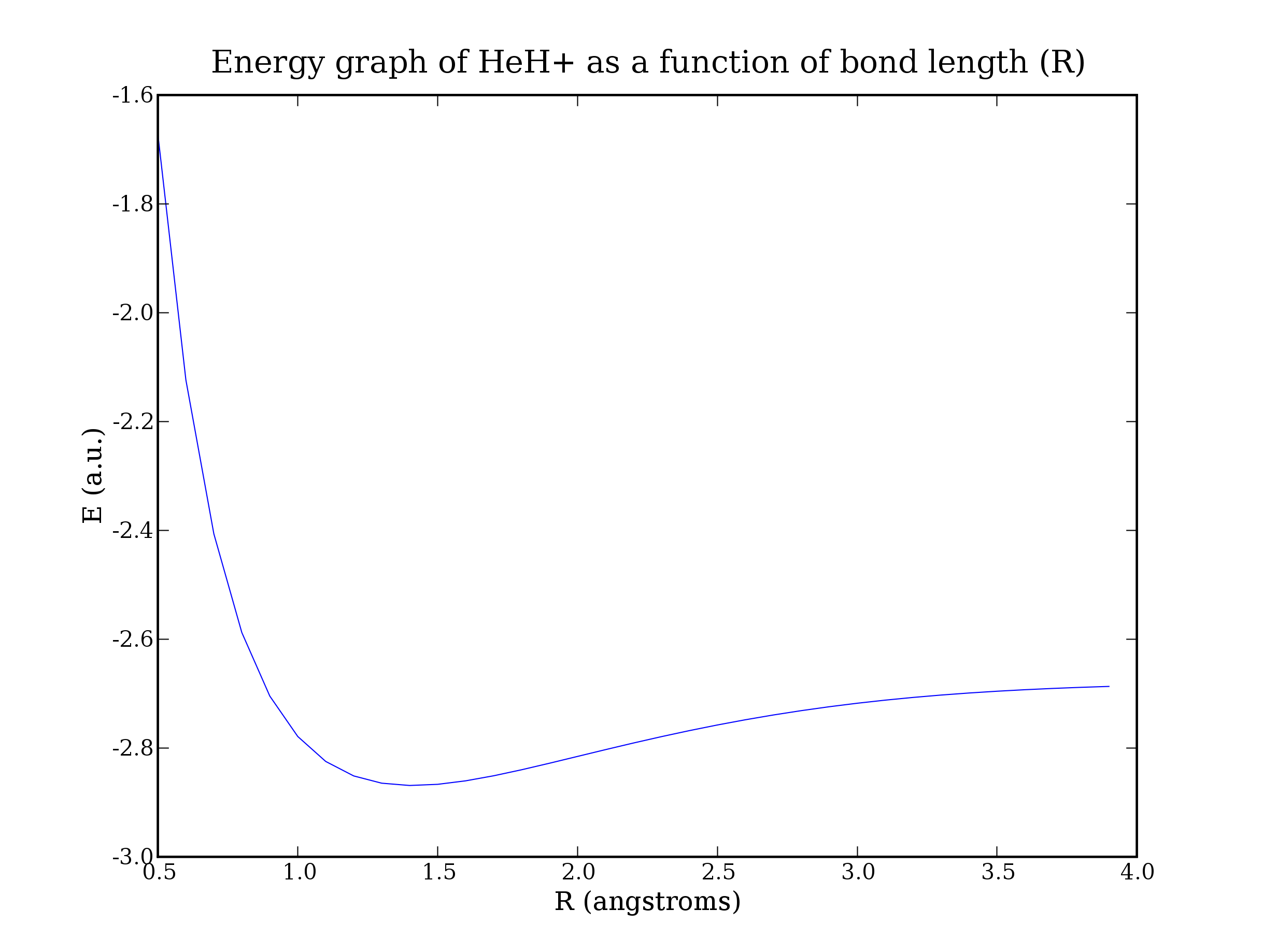
Using this procedure you can calculate the energy of a molecule in different configurations. Here is a calculation of HeH+. Varying the bond length and recalculating the energy can produce a graph or bond radius to energy - with the minimum point of the graph closely corresponding with experimental measurements of the true bond length.
 Furthermore, one can also visualize the electron density around the two nuclei by sampling the basis functions multiplies by their computed coefficients. The following image was produced by sampling this function over a grid - the two nuclear centres can easily be seen.
Furthermore, one can also visualize the electron density around the two nuclei by sampling the basis functions multiplies by their computed coefficients. The following image was produced by sampling this function over a grid - the two nuclear centres can easily be seen.

Finally, here is program output for performing a full calculation on HeH+ with the STO-3G basis set and a bond length of 1.4632.
----------- Overlap Matrix S(arb) -----------
[[ 1.00000143 0.46013279]
[ 0.46013279 1.00000134]]
----------- Kinetic Energy T -----------
[[ 2.06863805 0.17064196]
[ 0.17064196 0.76003235]]
----------- Nuclear Attraction V [ 0 ] -----------
[[-4.04729144 -1.10808097]
[-1.10808097 -1.26524563]]
----------- Nuclear Attraction V [ 1 ] -----------
[[-0.67645005 -0.42008591]
[-0.42008591 -1.22661494]]
----------- Core Hamiltonian Hcore -----------
[[-2.65510344 -1.35752492]
[-1.35752492 -1.73182821]]
----------- Electronic Repulsion Integrals -----------
( 1 1 | 1 1 ) = 1.27793341429
( 1 1 | 2 1 ) = 0.438653458227
( 1 1 | 2 2 ) = 0.604395613069
( 2 1 | 1 1 ) = 0.438653458227
( 2 1 | 2 1 ) = 0.182342598621
( 2 1 | 2 2 ) = 0.31802119604
( 2 2 | 1 1 ) = 0.604395613069
( 2 2 | 2 1 ) = 0.31802119604
( 2 2 | 2 2 ) = 0.774607905515
----------- Converging Electronic Energy -----------
Iteration 0 Electronic energy: 0.0
Iteration 1 Electronic energy: -4.15531813136
Iteration 2 Electronic energy: -4.23394307756
Iteration 3 Electronic energy: -4.23510236385
Iteration 4 Electronic energy: -4.23511168343
Converged at -4.23511168343 , Including Nuclear energy : -2.86824454292