# Since we set the `dataframe` parameter to `True`, the method returns our data as a nice and neat pandas dataframe. If set to `False`, the method returns a dictionary. We can all check to make sure all the columns are there by executing the `get_ephys_features` and `get_morphology_features` seperately and comparing the columns. # In[3]: all_features_columns = all_features.columns all_features_columns # In[4]: # Store all ephys columns in a variable ephys = pd.DataFrame(ctc.get_ephys_features()) ephys_columns = ephys.columns # Store all morphology columns in a variable morphology = pd.DataFrame(ctc.get_morphology_features()) morphology_columns = morphology.columns # Combine the two into one list ephys_and_morphology = list(morphology_columns) + list(ephys_columns) # Sort and Compare the columns to make sure they are all there print(list(all_features_columns).sort() == ephys_and_morphology.sort()) # By default, `get_all_features()` only returns ephys and morphology features for cells that have reconstructions. To access all cells regardless of reconstruction, set the parameter `require_recontruction` to `False`. # The `get_ephys_data()` method can download electrophysiology traces for a single cell in the database. This method returns a class instance with helper methods for retrieving stimulus and response traces out of an NWB file. In order to use this method, you must specify the id of the cell specimen whose electrophysiology you would like to download. # Below we have created a pandas dataframe from the data on human cells and set the row indices to be the `id` column. This will give us the metadata on human cells along and ID's to choose from. # In[5]:
# If we look back at our dataframe, our rows don't have any useful information -- they're simply a list of indices starting from zero. We can reassign the row values by using the method `set_index`. You can assign any present column in the dataframe as the indices to be the indeces for the rows. Since each observation in the dataframe has a unique `id`, the `id` will be set as the index.
# In[4]:
all_cells_df = all_cells_df.set_index('id')
all_cells_df.head()
# As you may have noticed already, out current dataframe only contains metadeta about our cells and no information on the morphology or electrophysiolgy of our cells. In order to get information about the morphology of these cells, we need to use the `get_morphology_features()` method on our instance of the cell types cache.
# In[5]:
#morphology = ctc.get_morphology_features()
# downloads the morphology features and sets up the dataframe all in one line
morphology_df = pd.DataFrame(
ctc.get_morphology_features()).set_index('specimen_id')
print('\nLength of dataframe:')
print(len(morphology_df))
morphology_df.head()
# Now we have two dataframes, one with the metadata for our cells (indexed by id) and another with the morphology data for all cells, also indexed by id. Usefully, these ids are unique to each cell, meaning we can match them across dataframes.
#
# We can use either the `merge` or `join` pandas methods in order to pull all of this data into one dataframe.
#
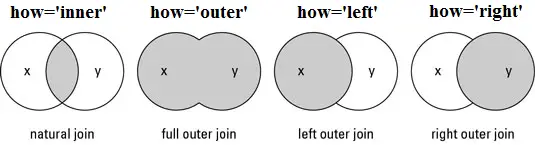
# 
#
# There are different types of joins/merges you can do in pandas, illustrated <a href="http://www.datasciencemadesimple.com/join-merge-data-frames-pandas-python/">above</a>. Here, we want to do an **inner** merge, where we're only keeping entries with indices that are in both dataframes. We could do this merge based on columns, alternatively.
#
# **Inner** is the default kind of join, so we do not need to specify it. And by default, join will use the 'left' dataframe, in other words, the dataframe that is executing the `join` method.
#
# If you need more information, look at the <a href="https://pandas.pydata.org/pandas-docs/stable/reference/api/pandas.DataFrame.join.html">join</a> and <a href="https://pandas.pydata.org/pandas-docs/stable/reference/api/pandas.DataFrame.merge.html">merge</a> documentation: you can use either of these to unite your dataframes, though join will be simpler!
def data_for_specimen_id(specimen_id,
sweep_qc_option,
data_source,
ap_window_length=0.005,
target_sampling_rate=50000):
logging.debug("specimen_id: {}".format(specimen_id))
# Find or retrieve NWB file and ancillary info and construct an AibsDataSet object
ontology = StimulusOntology(
ju.read(StimulusOntology.DEFAULT_STIMULUS_ONTOLOGY_FILE))
if data_source == "lims":
nwb_path, h5_path = lims_nwb_information(specimen_id)
if type(nwb_path) is dict and "error" in nwb_path:
logging.warning(
"Problem getting NWB file for specimen {:d} from LIMS".format(
specimen_id))
return nwb_path
try:
data_set = AibsDataSet(nwb_file=nwb_path,
h5_file=h5_path,
ontology=ontology)
except Exception as detail:
logging.warning(
"Exception when loading specimen {:d} from LIMS".format(
specimen_id))
logging.warning(detail)
return {
"error": {
"type": "dataset",
"details": traceback.format_exc(limit=None)
}
}
elif data_source == "sdk":
ctc = CellTypesCache()
morph = ctc.get_reconstruction(specimen_id)
morph_table = ctc.get_morphology_features(specimen_id)
morph_table.to_csv('cell_types\\specimen_' + str(specimen_id) + '\\' +
'morphology_features.csv')
print("morph dl failed")
nwb_path, sweep_info = sdk_nwb_information(specimen_id)
try:
data_set = AibsDataSet(nwb_file=nwb_path,
sweep_info=sweep_info,
ontology=ontology)
except Exception as detail:
logging.warning(
"Exception when loading specimen {:d} via Allen SDK".format(
specimen_id))
logging.warning(detail)
return {
"error": {
"type": "dataset",
"details": traceback.format_exc(limit=None)
}
}
else:
logging.error("invalid data source specified ({})".format(data_source))
# Identify and preprocess long square sweeps
try:
lsq_sweep_numbers = categorize_iclamp_sweeps(
data_set,
ontology.long_square_names,
sweep_qc_option=sweep_qc_option,
specimen_id=specimen_id)
(lsq_sweeps, lsq_features, lsq_start, lsq_end,
lsq_spx) = preprocess_long_square_sweeps(data_set, lsq_sweep_numbers)
except Exception as detail:
logging.warning(
"Exception when preprocessing long square sweeps from specimen {:d}"
.format(specimen_id))
logging.warning(detail)
return {
"error": {
"type": "sweep_table",
"details": traceback.format_exc(limit=None)
}
}
# Identify and preprocess short square sweeps
try:
ssq_sweep_numbers = categorize_iclamp_sweeps(
data_set,
ontology.short_square_names,
sweep_qc_option=sweep_qc_option,
specimen_id=specimen_id)
ssq_sweeps, ssq_features = preprocess_short_square_sweeps(
data_set, ssq_sweep_numbers)
except Exception as detail:
logging.warning(
"Exception when preprocessing short square sweeps from specimen {:d}"
.format(specimen_id))
logging.warning(detail)
return {
"error": {
"type": "sweep_table",
"details": traceback.format_exc(limit=None)
}
}
# Identify and preprocess ramp sweeps
try:
ramp_sweep_numbers = categorize_iclamp_sweeps(
data_set,
ontology.ramp_names,
sweep_qc_option=sweep_qc_option,
specimen_id=specimen_id)
ramp_sweeps, ramp_features = preprocess_ramp_sweeps(
data_set, ramp_sweep_numbers)
except Exception as detail:
logging.warning(
"Exception when preprocessing ramp sweeps from specimen {:d}".
format(specimen_id))
logging.warning(detail)
return {
"error": {
"type": "sweep_table",
"details": traceback.format_exc(limit=None)
}
}
# Calculate desired feature vectors
result = {}
(subthresh_hyperpol_dict, hyperpol_deflect_dict
) = fv.identify_subthreshold_hyperpol_with_amplitudes(
lsq_features, lsq_sweeps)
target_amps_for_step_subthresh = [-90, -70, -50, -30, -10]
result["step_subthresh"] = fv.step_subthreshold(
subthresh_hyperpol_dict,
target_amps_for_step_subthresh,
lsq_start,
lsq_end,
amp_tolerance=5)
result["subthresh_norm"] = fv.subthresh_norm(subthresh_hyperpol_dict,
hyperpol_deflect_dict,
lsq_start, lsq_end)
(subthresh_depol_dict,
depol_deflect_dict) = fv.identify_subthreshold_depol_with_amplitudes(
lsq_features, lsq_sweeps)
result["subthresh_depol_norm"] = fv.subthresh_depol_norm(
subthresh_depol_dict, depol_deflect_dict, lsq_start, lsq_end)
isi_sweep, isi_sweep_spike_info = fv.identify_sweep_for_isi_shape(
lsq_sweeps, lsq_features, lsq_end - lsq_start)
result["isi_shape"] = fv.isi_shape(isi_sweep, isi_sweep_spike_info,
lsq_end)
# Calculate waveforms from each type of sweep
spiking_ssq_sweep_list = [
ssq_sweeps.sweeps[swp_ind]
for swp_ind in ssq_features["common_amp_sweeps"].index
]
spiking_ssq_info_list = [
ssq_features["spikes_set"][swp_ind]
for swp_ind in ssq_features["common_amp_sweeps"].index
]
ssq_ap_v, ssq_ap_dv = fv.first_ap_vectors(
spiking_ssq_sweep_list,
spiking_ssq_info_list,
target_sampling_rate=target_sampling_rate,
window_length=ap_window_length,
skip_clipped=True)
rheo_ind = lsq_features["rheobase_sweep"].name
sweep = lsq_sweeps.sweeps[rheo_ind]
lsq_ap_v, lsq_ap_dv = fv.first_ap_vectors(
[sweep], [lsq_features["spikes_set"][rheo_ind]],
target_sampling_rate=target_sampling_rate,
window_length=ap_window_length)
spiking_ramp_sweep_list = [
ramp_sweeps.sweeps[swp_ind]
for swp_ind in ramp_features["spiking_sweeps"].index
]
spiking_ramp_info_list = [
ramp_features["spikes_set"][swp_ind]
for swp_ind in ramp_features["spiking_sweeps"].index
]
ramp_ap_v, ramp_ap_dv = fv.first_ap_vectors(
spiking_ramp_sweep_list,
spiking_ramp_info_list,
target_sampling_rate=target_sampling_rate,
window_length=ap_window_length,
skip_clipped=True)
# Combine so that differences can be assessed by analyses like sPCA
result["first_ap_v"] = np.hstack([ssq_ap_v, lsq_ap_v, ramp_ap_v])
result["first_ap_dv"] = np.hstack([ssq_ap_dv, lsq_ap_dv, ramp_ap_dv])
target_amplitudes = np.arange(0, 120, 20)
supra_info_list = fv.identify_suprathreshold_spike_info(lsq_features,
target_amplitudes,
shift=10)
result["psth"] = fv.psth_vector(supra_info_list, lsq_start, lsq_end)
result["inst_freq"] = fv.inst_freq_vector(supra_info_list, lsq_start,
lsq_end)
spike_feature_list = [
"upstroke_downstroke_ratio",
"peak_v",
"fast_trough_v",
"threshold_v",
"width",
]
for feature in spike_feature_list:
result["spiking_" + feature] = fv.spike_feature_vector(
feature, supra_info_list, lsq_start, lsq_end)
return result
